2. AstroLogics Tutorial, Boolean Network Analysis and Clustering : Herault Hematopoiesis¶
This tutorial demonstrates the AstroLogics framework for analyzing and comparing Boolean network model ensembles. AstroLogics is designed for benchmarking Boolean models through three major evaluation criteria: network evaluation, logical function evaluation, and dynamic evaluation.
2.1. Overview of AstroLogics Framework¶
AstroLogics addresses a critical gap in Boolean network modeling: while multiple methods exist for Boolean model synthesis (like Bonesis, BN-sketch), there hasn’t been a standardized way to evaluate and compare generated model ensembles. The framework focuses on:
Dynamic properties: Examining state transition graphs and model behaviors through simulation
Logical function evaluation: Analyzing and comparing logical rules that govern node behaviors
Model clustering: Identifying groups of models with similar dynamics and logical features
2.2. Required Libraries¶
[ ]:
import pandas as pd
import os
import astrologics as ast
import seaborn as sns
import matplotlib.pyplot as plt
2.2.1. Dataset: Herault hematopoeisis model¶
We’ll use the Herault Hematopoiesis model from Herault et al., 2018, which provides an excellent framework for studying blood cell differentiation. This model contains 33 nodes representing key transcription factors and signaling molecules involved in hematopoietic lineage specification. The model demonstrates hierarchical decision-making processes during blood cell development from hematopoietic stem cells to differentiated blood cell types.
2.3. Step 1: Load Model Ensemble¶
[ ]:
model_path = '../models/herault_hematopoiesis/'
model = ast.ensemble(model_path, project_name = 'herault_hematopoiesis')
model.create_simulation()
Simulation object created
The ensemble object is the core component of AstroLogics that handles:
Loading multiple Boolean network models from a directory
Managing simulation parameters and configurations
Coordinating analysis across the model ensemble
2.4. Step 2: Simulate the model ensemble¶
In this part of the script we first simulate all the BN within the model ensemble. We utilize the MaBoSS engine as the main simulator.
This creates an initial state where all nodes have equal probability (0.5) of being active. This represents a neutral starting condition that allows the system to evolve according to its inherent dynamics.
We then start the simulation using MaBoSS.
MaBoSS (Markovian Boolean Stochastic Simulator) is crucial for the AstroLogics approach because:
It converts Boolean network dynamics into continuous-time Markov processes
Provides probabilistic approximation of complex state transition graphs
Enables analysis of both transient and steady-state behaviors
Scales computationally better than exhaustive state space exploration
[ ]:
# Configure simulation parameters
model.simulation.update_parameters(max_time = 20,thread_count = 15, sample_count = 2000)
model.simulation.run_simulation()
Start simulation
Simulation completed
2.5. Step 3 : Calculate the trajectory and visualize the distance matrix using MDS¶
In this part of the script, we compare the two method of calculating the distance between models.
In this function calculate_distancematrix, users can select the two options of data used to calculate the distance.
endpoint: The endpoint utilize the node activation probability at the endpoint of MaBoSS simulation. User could also defines thetimepointto define a specific timepoint they want to use to define the distancetrajectory: This options utilize the whole MaBoSS simulation trajectory and thedtwmethod to calculate the distances between models.
In this part, we show different results that could be obtained from two different method.
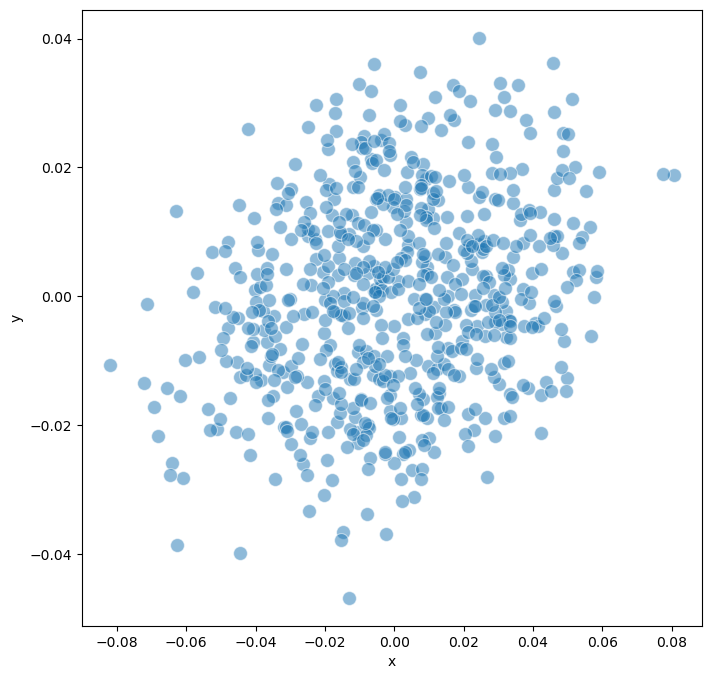
Using
endpointwe showed no clear separation of the model, which coresspond to the single attractors that all the model could reach.On the other hand, when using
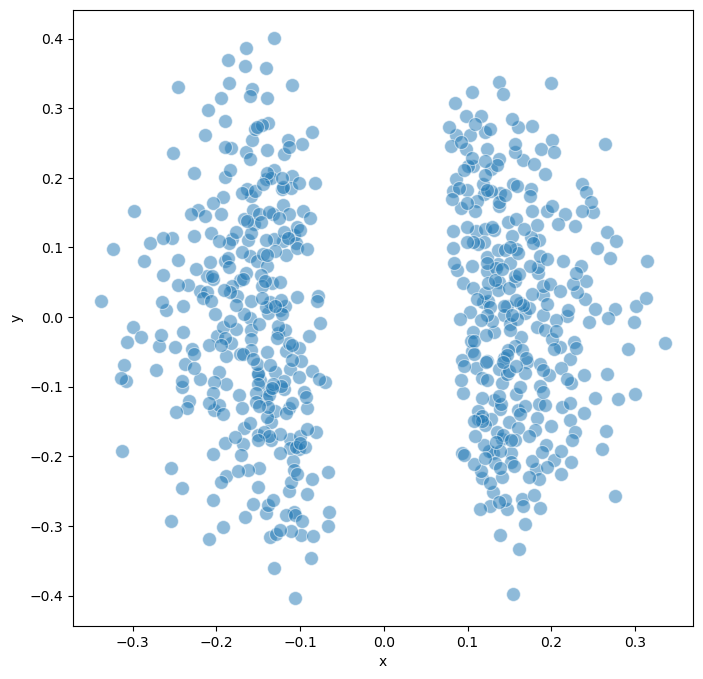
trajectorywe can observe two major cluster of models based on the different signaling path that two groups of BNs took.
[5]:
model.create_trajectory()
Trajectory object created
[6]:
model.trajectory.calculate_distancematrix(mode = 'endpoint')
# Perform MDS (Multidimensional Scaling) for visualization
model.trajectory.calculate_MDS()
model.trajectory.plot_MDS(s = 100, fig_size = (8,8))
Calculating distance matrix for endpoint simulation...
Distance matrix calculated successfully.
[7]:
model.trajectory.calculate_distancematrix(mode = 'trajectory')
# Perform MDS (Multidimensional Scaling) for visualization
model.trajectory.calculate_MDS()
model.trajectory.plot_MDS(s = 100, fig_size = (8,8))
Calculating distance matrix for whole trajectory...
Distance matrix calculated successfully.
2.6. Step 4: Model Clustering¶
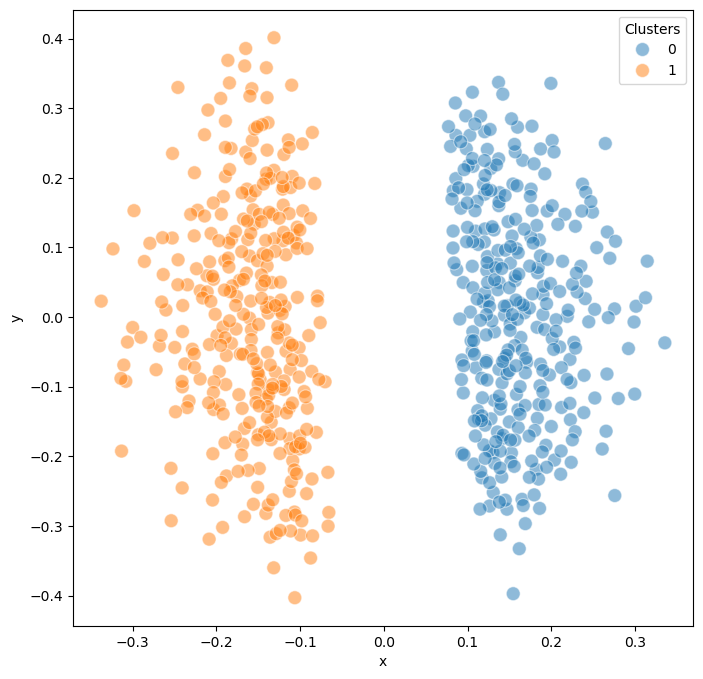
Clustering reveals distinct groups within the model ensemble. In this example, we found 2 major clusters corresponding to different attractor groups, representing distinct cellular fate decisions.
[8]:
model.trajectory.calculate_kmean_cluster(n_cluster = 2,
random_state = 0)
Calculated k-means clustering with 2 clusters.
[9]:
model.trajectory.plot_MDS(s = 100, fig_size = (8,8),plot_cluster = True)
2.7. Step 5: Logic Function Analysis¶
This step implements the logical function evaluation component of AstroLogics:
Converts Boolean equations to Disjunctive Normal Form (DNF)
Creates feature matrices comparing logical rules across models
Identifies constant, varied, and marker clauses
[10]:
model.create_logic()
model.logic.model_logic
model.logic.create_flattend_logic_clause()
Loading models logics
Concatenate results into matrix
Logic object created
Flatten models logic clauses
Concatenate results into matrix
Flattend logic clause created
2.8. Step 6 : Calculate statistic of Logic features (clauses)¶
In this steps, we have already featurized the logical equations into model logics or clauses.
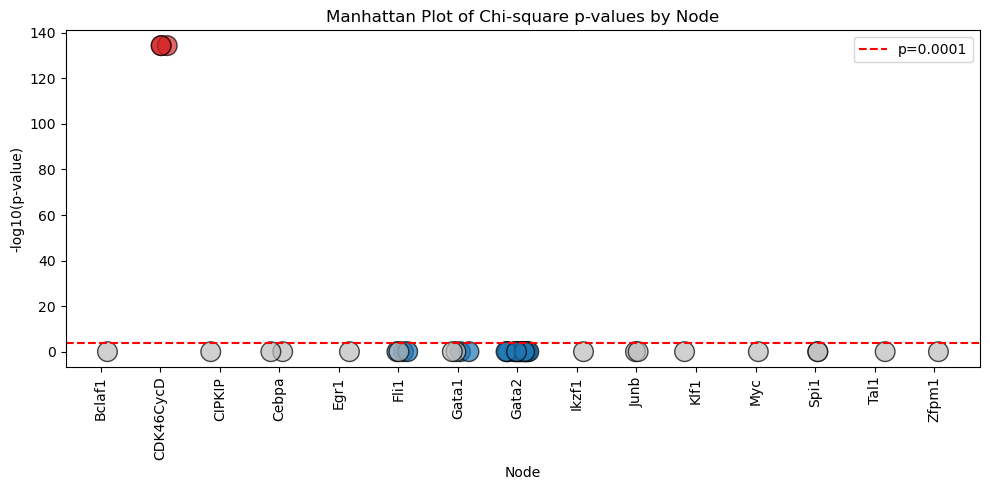
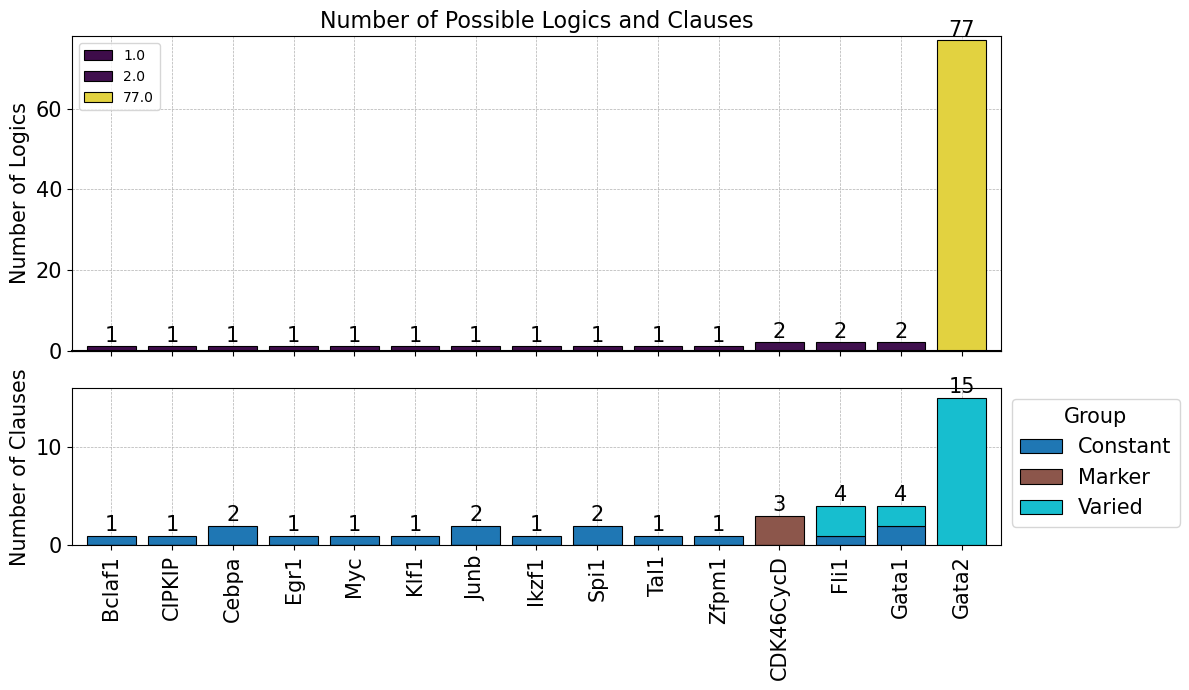
We can then integrate the clusters obtained from the trajectory analysis into the .logic and perform chi-square statistical test to categorize logic features (clauses) into 3 major groups
Constatnt : core regulatory features that appears across BNs in the model ensemble
Varied : Features that may differ between individual BNs but show no statistical significant
Marker : Key discriminatory features that statistically distinguish between different model clusters.
We can define the p-value of the chi-square test using the function pval_threshold.
[11]:
model.logic.map_model_clusters(model.trajectory.cluster_dict)
model.logic.calculate_logic_statistic(pval_threshold = 0.0001)
Model clusters mapped to logic clauses
The results of the analysis can be visualized in the form of Manhattan Plot shown below.
[12]:
model.logic.plot_manhattan()
Or the results can be summarized into the barplot shown here
[13]:
model.logic.plot_logicstat_summary()
2.9. Step 7: Advanced Trajectory Analysis¶
These visualizations help identify:
Most variable nodes: Components showing greatest differences between models
Critical regulators: Nodes whose activity patterns distinguish model clusters
Temporal patterns: How specific nodes behave over simulation time
In this first plot, we check what are the features that shows the highest variance in their dynamics accross simulation. We calculate the variance of node activation probabily of all BNs in the model ensemble across all timepoints and plotted using the heatmap.
[14]:
model.trajectory.plot_trajectory_variance()
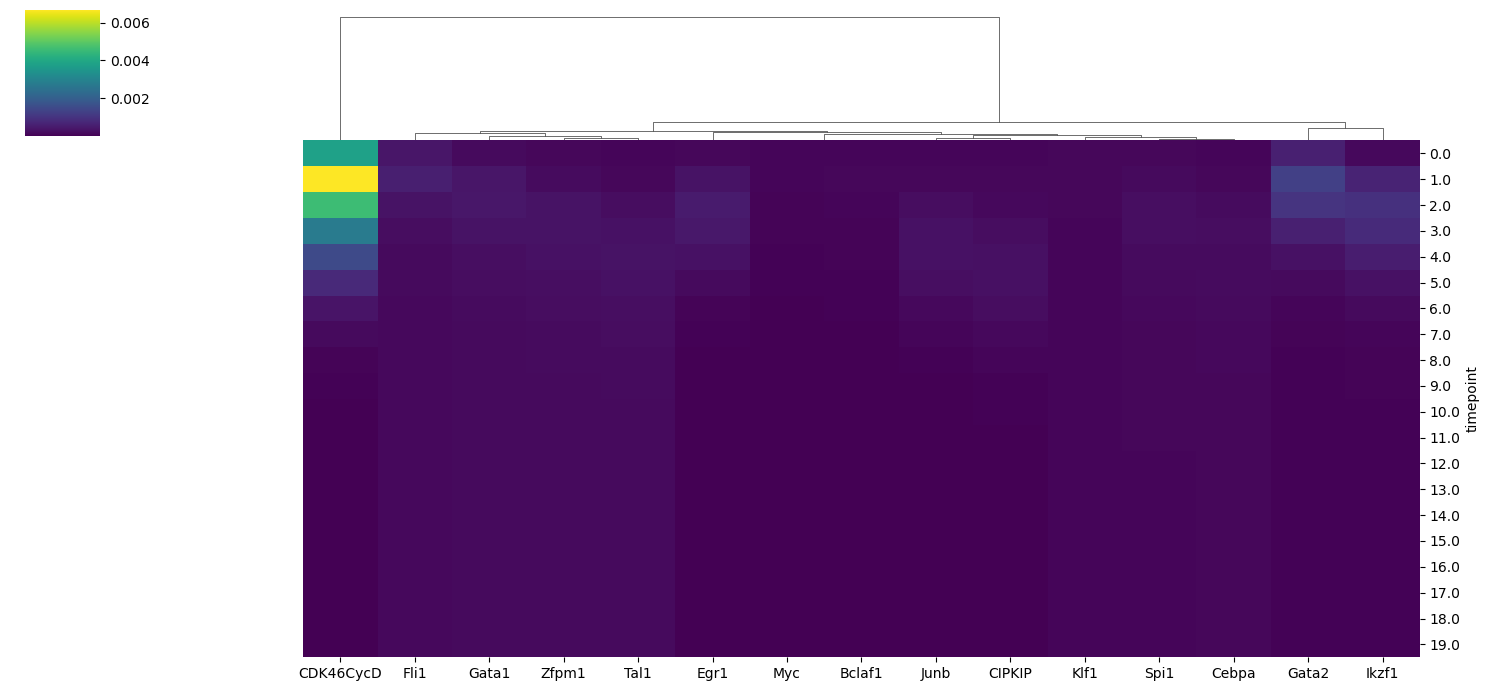
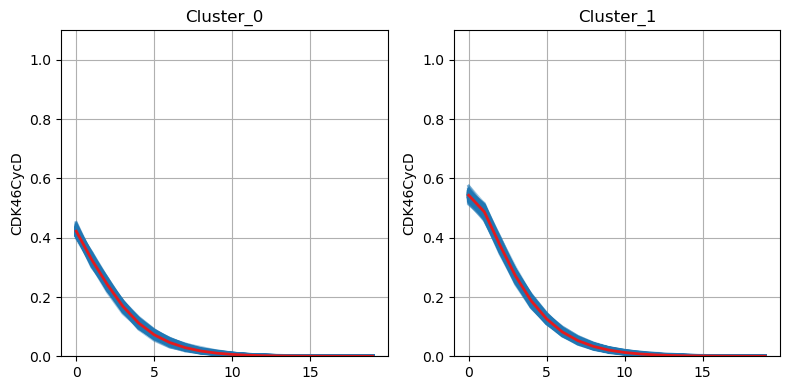
We identify the key node of interest (CDK46CycD) which shows the highest variance along the timepoints. Finally, we could visualize the dynamics of this node between two identified clusters using the lineplot.
From this, we found that the dynamics of this node differ during the early timepoint of the simulation, while finally converge to 0 at the later timepoint.
[15]:
model.trajectory.plot_node_trajectory(node = ['CDK46CycD'])
2.10. Step 8 : Logic Feature Heatmap¶
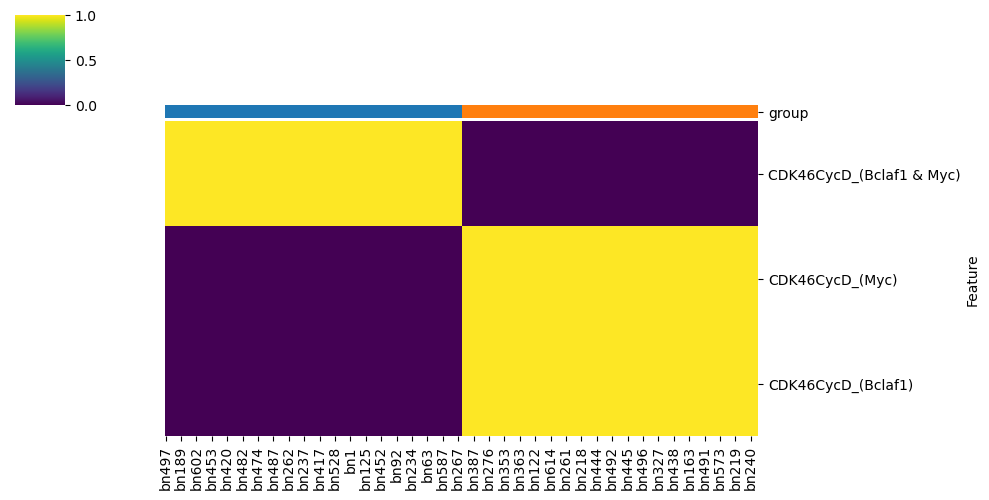
This analysis typically reveals that Myt1L is the key distinguishing node between clusters:
Cluster 0: CDK46CycD is regulated by Bclaf1 & Myc
Cluster 1: CDK46CycD is regulated by Bclaf1 | Myc
[16]:
model.logic.plot_node_logic_heatmap(node = ['CDK46CycD'],
fig_size = (8, 8))
<Figure size 800x800 with 0 Axes>